Thanks to a new medicine, hydoxyurea, sickle cell sufferers like Rod now have a better chance of living an almost normal life, with fewer painful attacks and hospitalizations.
Rod's Medical Story
Rod's childhood was mostly uneventful, although he had many episodes of aches and pains as he was growing up. At 18, he was hospitalized with his first "sickle cell crisis," where red cells broke down and blocked blood flow, especially in tiny arteries throughout Rod's body. Rod was even treated with exchange transfusion on one occasion when his lungs were plugged up with misshapen sickle cells and his life was in danger — we replaced his defective red blood cells with normal red blood cells. Exchange transfusion is not an ordinary part of medical care for crisis but, sometimes, it can be life saving when a patient has an acute chest syndrome like Rod did. Since then, he's had many crises and many medical problems, including episodes of severe abdominal and back pains, skin ulcers and bone and joint damage.
What is Sickle Cell Disease?
Sickle cell anemia is the most clinically significant of a group of diseases in which sickle hemoglobin is present. It is so named because the normal round shape of the red blood cells is, instead, sickle shaped. It was first described by James Herrick 90 years ago.
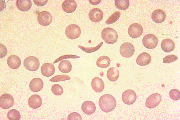
Figure 1. Three views of the peripheral blood smear of sickle cell anemia with red blood cell showing typical sickle shape.
From the American Society of Hematology Slide Bank. Used with permission.
From the American Society of Hematology Slide Bank. Used with permission.
The Nobel Prize chemist, Linus Pauling, deduced the genetic nature of the disease. Hemoglobin molecules in normal red blood cells bind oxygen in the lung for delivery to the body's cells. Sickle cells have a genetically defective hemoglobin and, although they bind oxygen, upon its release and delivery to the body's cell, the remaining hemoglobin, now de-oxygenated, crystallizes. The crystals distort the shape of the red blood cell giving it the sickle appearance. The disease primarily affects Africans and those of African heritage. It has been suggested that the sickle cell mutation thrived because it protected people from a more deadly disease, malaria. An online database cataloging human genetic diseases, including all the hemoglobin diseases, is available at the National Center for Biotechnical Information.
The Current Treatment
My patient's medical experience brings up several issues about the treatment of sickle cell disease. The new medicine, hydroxyurea, does not cure the disease but it can reduce the number of "crises." Unfortunately, for Rod and other sickle cell disease carriers, the crises will continue to occur, usually unpredictably, and can result in hospitalizations which average five to seven days duration. There is no specific therapy for a crisis — treatment is pain management, hydration and treatment of complications, if present.
In every hospital where I have worked, patients are, unfortunately, too often given inadequate and infrequent doses of meperidine, a narcotic pain killer. Many sickle cell anemia patients have life-long experience with this approach and state their expectations when arriving at emergency departments. They may ask to be started on 100 to 200 mg of meperidine, only to find that the emergency department staffers believe that they are addicts and start them with lower doses.
Meperidine is not recommended as a first line narcotic for a sickle cell crisis because it needs to be given every two hours and a form of meperidine, chemically changed by the body, can accumulate to toxic levels, causing agitation and even seizures. When narcotics are needed, morphine or hydromorphone are better choices. The Agency for Health Care Policy and Research has developed pain management guidelines that are very useful. They should be adjusted according to the patient's past experience....[P]atients are, unfortunately, too often given inadequate and infrequent doses of meperidine, a narcotic pain killer.
A sickle cell crisis occurs when the defective red blood cells clump and block arteries, interrupting blood supply to important organs. Exchange transfusion therapy is particularly effective. Using pheresis machines, now widely available, doctors can carry out the transfusion quickly. Chronic transfusion therapy can reduce the frequency of these painful and serious complications (stroke, heart damage, lung infection) but carries the risks of iron overload, blood borne illness and the other problems of transfusion dependence.
Newer alternatives, such as bone marrow transplantation, can cure sickle cell disease but they can also cause early death, as well as graft versus host disease, cancer, sterility, neurologic and other complications.
The new medicine, hydroxyurea, which is most easily taken orally, markedly reduces the frequency of sickle cell crisis. Hydroxyurea is one of several agents that can raise fetal hemoglobin levels in sickle cell anemia patients. Fetal hemoglobin, which, as its name implies, carries our oxygen before we are born, is free of the defective hemoglobin. Large-scale clinical trials established that hydroxyurea is clinically effective in decreasing the incidence of crises.6 There are some doubts about whether the rise in fetal hemoglobin really explains its clinical benefits because the data did not show that the decrease in incidence of crises correlated with the increase in fetal hemoglobin. In fact, the decrease in crises occurred even before the fetal hemoglobin rose.
Despite these caveats, the results of the hydroxyurea trial were so striking that an alert was sent over the Internet to Medline® users by the NIH when the data were first released.
Many unknowns are ahead of us but it is very gratifying to be able, finally, to offer some effective therapy for sickle cell anemia.