
Hard and tight skin, most often on the face and hands, is the most prominent feature of scleroderma. Also known as Systematic Sclerosis (SSc) and sometimes characterized as the "disease that turns people into stone," scleroderma is especially frightening because we do not know what causes it.
The word "scleroderma" comes from two ancient Greek words: "sclero," meaning hard, and "derma," meaning skin. Yet despite its name, scleroderma can also damage internal tissues and organs and it is this damage which is far more dangerous to a person's health than changes in the skin.
On the positive side, medical researchers have made progress. Better treatments are now available for some of the symptoms of SSc. And, the disease does not appear to be contagious, infectious, cancerous or malignant in any way.
Thankfully, SSc is also rare — affecting approximately 300,000 persons in the United States — although not as rare as better known diseases such as ALS, or Lou Gehrig's Disease, which affects about 30,000 Americans in an average year.Uncertainty and frustration take a toll on the psychological well-being of SSc sufferers...
Uncertainty and frustration take a toll on the psychological well-being of SSc sufferers, who often describe the period before they are diagnosed as the most difficult part of the illness. To give an idea of the vagueness and complexity of SSc, in 1980 the American College of Rheumatology defined it as the presence of a specific type of skin hardening near the metacarpal phalangeal (MCP) joints of the hand, or two of the following:
- sclerodactyly, or stiffness and tightness of the skin of the fingers
- scarring of the fingertips
- loss of fat pad tissue on the fingers
- pulmonary fibrosis, or the growth of excess fiber tissue in the lungs
- Diffuse Cutaneous SSc, or dcSSc
- CREST, or Limited Cutaneous SSc. CREST is an acronym that stands for its telltale symptoms: Calcinosis, Raynaud's Syndrome, Esophageal dysmotility, Sclerodactyly, and Telangectasias. Calcinosis is the presence of calcium deposits, usually in the fingers; Raynaud's is a sudden loss of blood circulation to the extremities, often in response to cold; Esophageal dysmotility is a loss of muscle control of the esophagus, which can interfere with swallowing; Sclerodactyly is a tapering deformity of the bones of the fingers; and Telangiectasias are numerous, small red spots on the skin of the fingers and face, or on the inside of the mouth.
- Sine Scleroderma
- Scleroderma with an Overlap Syndrome.
The main feature that distinguishes dcSSc from CREST is the extent to which it affects the skin. While both cause skin thickening on the hands and face, dcSSc causes skin thickness on the upper arms, thighs and torso as well. (Figures 1, 2).
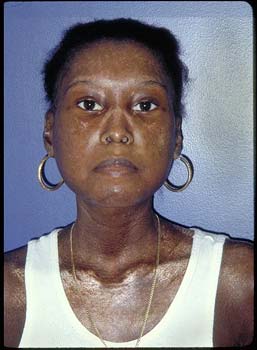
Note the taught, shiny skin over the face and chest, and how the lips have lighter pigmentation (hypopigmentation) and are pursed.
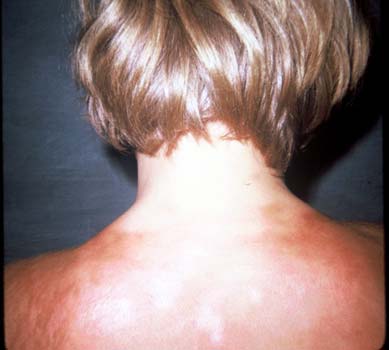
Note the extensive skin changes on the back.
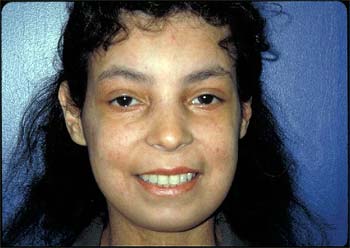
Along with tightened, shiny skin over the face, there are widespread telangectasias, or dilated blood vessels, creating numerous red spots on the skin, and a tightened mouth opening.
The two types of scleroderma are different in other ways as well. DcSSc tends to be a more aggressive disease, with more and stronger symptoms early on. The typical first sign of dcSSc is Raynaud's Syndrome, followed within a year by skin thickening, especially of the hands; overall fatigue; and joint problems. People suffering from dcSSc are more likely than those with CREST to go on to develop lung, kidney, gastrointestinal and heart problems. CREST progresses more slowly; sufferers sometimes have Raynaud's for many years before any skin changes. They are also far less likely ever to develop internal organ problems.
This difference in how they affect the body's internal organs explains the great difference in mortality rates between the two types of SSc. There is a five-year mortality rate of approximately 30-40% for dcSSc, as compared to only 20% for CREST.DcSSc tends to be a more aggressive disease, with more and stronger symptoms early on.
Understandably, the first reaction of those who are told that they have this disease is: "Why me?" That is an important question, one which medical science is vigorously trying to answer. Some research has suggested possible genetic causes or predispositions for SSc. Interestingly, however, studies of twins have not shed any light on this question.
The research on SSc echoes a recurring theme in autoimmune diseases — that while genetic susceptibility may be a prerequisite for the disease, it is probably not the only factor in determining whether or not someone gets the disease.
Most researchers feel that SSc is triggered by some environmental insult to the system of someone who is already susceptible, whether for genetic or other reasons. What kind of insult is not clear, despite epidemiological studies that have implicated exposure to some types of chemical solvents, silica dust and silicone implants. The fact that geographic "clusters" of SSc have been identified in England, in Italy and among the Choctaw Indians in Oklahoma provides strong evidence for the "environmental trigger" theory.
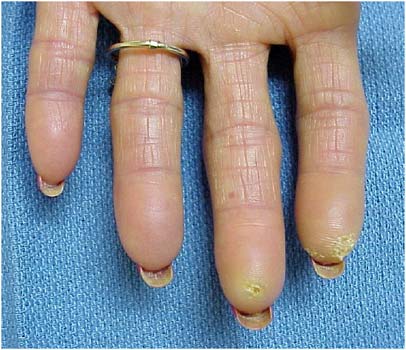
Note how the fingertips have lost their fat pads and appear pitted.
The good news for those with SSc and kidney problems is that Scleroderma renal crisis, a serious kidney condition, is now very treatable with a class of drugs known as ACE inhibitors.
When dcSSc affects the lungs, it does so in three ways, by causing what is known as interstitial lung disease (ILD) with fibrosis (22%), pulmonary hypertension (19%), or both. Pulmonary hypertension is a blood vessel disorder in the lung that produces shortness of breath, chest pain, dizzy spells and fainting.It is inevitable that most SSc sufferers will have gastrointestinal problems at some point. Virtually all will develop reflux disease...
It is inevitable that most SSc sufferers will have gastrointestinal problems at some point. Virtually all will develop reflux disease, which causes stomach acid to regurgitate up into the throat. 8% of those with dcSSc will go on to develop more severe pseudo-obstruction and malabsorption conditions of the large or small intestines. These can progress to the point where the victims can no longer eat. Myocardial fibrosis, or the development of excess fibrous tissue around the heart, occurs in 15% of dcSSc patients.
Because people with CREST have fewer and less severe internal organ problems than those with dcSSc, they have a better overall outcome. CREST often progresses slowly and with subtle skin changes that are preceded for years by Raynaud's. Renal crisis and serious lung disease are virtually unheard of in CREST. Esophageal reflux disease is common in CREST but severe gastrointestinal problems such as malabsorption are not. Long-standing Raynaud's, telangectasias and calcinosis are classic hallmarks of the disease. Figures 3 and 5 demonstrate the facial telangectasias characteristic of CREST.
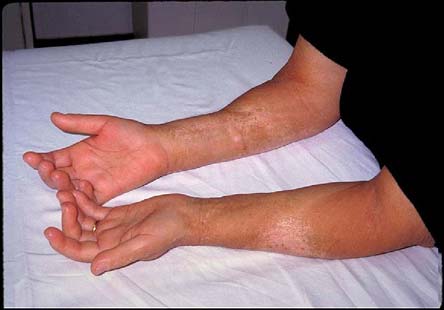
The skin over the hands and forearms is very tight and thickened.
Finally, not surprisingly, depression is common in people with SSc, occurring in about half of all cases.
Pulmonary hypertension is extremely difficult to treat. Infusions of the drugs iloprost or epoprostanol can be effective but they are prohibitively expensive and difficult to administer. Calcium channel blocker drugs are somewhat useful, but only in a minority of cases. Test results for the recently approved drug Bosentan®, however, are extremely promising.
Despite much effort and research, there are unfortunately no good treatments for skin thickening. The only reassurance for those with dcSSc is that the worst will be over after about two years.
The best treatment for chronic reflux disease is the use of proton pump inhibitor drugs, which block the body's production of stomach acid. There are currently no good treatments for small intestine malabsorption caused by SSc, other than, if it comes to that, providing nutrition through a feeding tube or IV.
Raynaud's Syndrome is another difficult to treat SSc symptom. Calcium channel blocker drugs help only a little with Raynaud's. Iloprost does seem to help reduce the frequency and severity of Raynaud's attacks.




